Генетика о паталогиях
Сообщений 1 страница 20 из 27
Поделиться216.08.2011 15:14
Наследственные хромосомные стоматологические заболевания
Введение.
Одним из разделов наследственной патологии (соответствующие больные занимают почти 25 % коечного фонда всего мира) являются хромосомные болезни. К ним можно отнести группу болезней, вызываемых числовыми или структурными изменениями хромосом либо их сочетанием, что обнаруживается при специальном анализе ядер клеток — кариологическом исследовании.
Черепно- лицевые аномалии, в частности морфологические изменения в зубах, могут быть обусловлены хромосомными аберрациями, генной мутацией, а так же совместными действиями многих генов и факторов среды. такие мультифакторные заболевания являются распространенной группой наследственных заболеваний и врожденных пороков развития.
Различные симптомы и болезни, при которых поражается черепно- лицевая область, нередко ассоциируется с изменениями в других органах и системах организма. Следовательно, для современной диагностики, профилактики и лечения необходимо сотрудничество клиницистов различного профиля и генетиков. Стоматологу- педиатору, ортодонту очень важно знать стоматологические проявления наследственных болезней и синдромов. Раннее их выявление совместно с педиатором, генетиком необходимо для определения прогноза и выбора правильного метода лечения.
1. Хромосомы и хромосомные болезни.
У высших организмов связь поколений осуществляется через половые клетки. Клетка — единое целое, и все ее структурные и биохимические компоненты тесно взаимосвязаны между собой. Еще в начале нашего века было установлено, что клетка имеет высокоспециализированные структурные элементы, которые определяют наследственную преемственность свойств организма. Этими элементами являются хромосомы (от греческого слова «хромое» — красящийся), которые включают в себя единицы наследственной информации — гены. Таким образом, каждая клетка является хранителем наследственной информации. Клетка имеет цитоплазму и ядро. Функции хранения и передачи наследственной информации в основном связаны с хромосомами клеточного ядра. Информация, содержащаяся в хромосомах оплодотворенного яйца, во время индивидуального развития должна быть передана всем клеткам тела. Передача информации от материнской клетки дочерним осуществляется во время клеточного деления при активном участии ядра и цитоплазмы. Специфическое значение в точном распределении хромосом между дочерними клетками принадлежит центросоме и митотическому аппарату клетки.
Для каждого биологического вида характерно постоянное число хромосом. У большинства высших организмов каждая клетка содержит диплоидный (2п) хромосомный набор. Хромосомы отличаются друг от друга формой и размерами. Совокупность количественных и качественных признаков хромосом, определяемая при микроскопировании в единичной клетке, называется кариотипом.
Нормальное диплоидное число хромосом у человека равно 46. Из-за несовершенства цитологической техники общее число хромосом у человека долго (с 1912 по 1956 г.) считали равным 48. В 1956 г. шведские цитологи J. H. Tijo и A. Levan применив усовершенствованную цитологическую методику, на материале культуры фибро-бластов легочной ткани 4 человеческих эмбрионов показали, что модельное число хромосом у человека равно 46. Эти данные в том же году были подтверждены английскими цитологами С. Е. Ford и J. L. Hamerton (1956). Эти два сообщения стали началом бурного развития цитогенетики человека.
Среди многих методов изучения наследственной патологии цитогенетический метод занимает важное место. С его помощью можно провести анализ материальных основ наследственности и кариотипа человека в норме и при патологии, изучить некоторые закономерности мутационного и эволюционного процессов. Все хромосомные болезни у человека были открыты этим методом. Он незаменим для дифференциальной диагностики многих врожденных и наследственных болезней. Овладеть им в условиях клинической лаборатории с соответствующей аппаратурой и реактивами несложно.
Кариотип человека определяется 46 хромосомами. Это число хромосом содержится в соматических клетках, половые клетки имеют набор в 2 раза меньший — 23 хромосомы. Из 46 хромосом человека 22 пары одинаковы у мужчин и женщин, их называют аутосомами. Они имеют порядковый номер от 1-го (самая крупная с центромерой в середине) до 22-го (самая маленькая с центромерой у края). В 23-й паре имеется отчетливая половая дифференцировка: в клетках тела у женщин находятся две крупные вполне идентичные друг другу хромосомы X, у мужчин имеется только одна хромосома X, а ее партнером служит маленькая хромосома У. Хромосомы Х и У называют половыми хромосомами.
При цитогентическом исследовании для того, чтобы ответить на вопрос, нормален ли хромосомный набор или имеется какая-либо аномалия, существенное значение приобретает правильный отбор метафазных пластинок. Для этого необходимы следующие условия: цельность метафазной пластинки; отсутствие или небольшое число взаимных наложений хромосом, средняя степень их конденсации (спирализации); обособленность метафазных пластинок друг от друга. Соблюдение этих правил позволяет в целом провести правильную идентификацию хромосом. Хромосомный анализ проводят в несколько этапов: визуальный анализ хромосомных препаратов; анализ хромосом с помощью зарисовки; анализ хромосом с помощью фотосъемки и раскладки кариотипа. Данные цитогенетических исследований заносят в специальные бланки — протоколы.
Из всех 23 пар хромосом с помощью рутинного метода можно идентифицировать только хромосомы 1; 2; 3;16 и У. Остальные хромосомы трудно различимы. Именно невозможность идентификации каждой хромосомы с помощью рутинного метода существенно ограничивала цитогенетическую диагностику и классификацию хромосомных болезней. Только с освоением новых методических подходов к изучению хромосом удалось, наконец, решить этот вопрос.
Линейная исчерченность хромосом выявляется после воздействия на них некоторых солевых растворов со строго заданным значением рН и определенным температурным режимом и с последующей окраской флюоресцирующими (Q-окраска) или основными красителями типа раствора Гимзы (G- и С-окраска). Помимо указанных способов окраски хромосом, применяют и другие специфические методы, которые позволяют избирательно окрашивать участки тех или иных хромосомных районов.
Наиболее информативным из них является метод С-окраски, который позволяет выявлять плотнокрасящи-еся сегменты, расположенные в центромерных или около-центромерных участках всех хромосом, а также в коротких плечах хромосом 13—15; 21—22 и в длинном плече хромосомы Y. С помощью этого метода обнаруживается так называемый структурный гетерохроматин. Значение метода С-окраски состоит в том, что он, выявляя структурный гетерохроматин во всех хромосомах, позволяет лучше, чем какой-либо другой метод, оценивать хромосомный полиморфизм у человека, т. е. межиндивидуальные различия по отдельным хромосомам. Для полиморфизма хромосом человека характерны наличие определенного варианта строения хромосомы во всех клетках, его передача от родителей к детям как простого моногенного признака, отсутствие заметного фенотипического эффекта. Уже твердо установлено, что истинный полиморфизм хромосом обусловлен вариабельностью в размерах их гетерохроматиновых районов.
Нормальная изменчивость, ранее обнаруживаемая лишь для немногих хромосом набора и у отдельных индивидов, на самом деле явление, широко распространенное. У каждого индивида оно проявляется специфическим сочетанием вариантов хромосом, и неограниченное число подобных сочетаний обеспечивает уникальность кариотипа каждого человека.
Использование новых методов современной генетики и генной инженерии позволило медицинским генетикам выявлять и клонировать участки хромосомной ДНК, .отвечающие за проявление наследственных дефектов, и использовать их в качестве основного материала в пренатальной диагностике.
Рассмотрим проблему пола в плане цитогенетики более подробно. В 1949 г. М. L. Вагг и Е. С. Вег при изучении клеток животных установили генетическую разницу между полами. В 1954 г. К. L. Moore и М. L. Вагг эту генетическую особенность подтвердили, исследуя клетки человека. Были обнаружены два типа клеток. В ядрах соматических клеток нормальной женщины была выявлена компактная хроматиновая глыбка, названная половым хроматином, или тельцем Барра, а в ядрах клеток нормального мужчины такая глыбка отсутствовала. Впоследствии установили, что обнаруженное тельце представляет собой неактивную хромосому .X. Тельце Барра чаще всего располагается на периферии у ядерной мембраны и его форма варьирует от треугольной до выпуклой. Для выявления полового хроматина обычно применяют анализ эпителиальных клеток в соскобе слизистой оболочки щеки. Наличие или отсутствие тельца Барра характеризует набор хромосом X, а следовательно, и пол индивида. Оказалось, что тельце Барра образуется из одной хромосомы X. Поэтому у женщин обнаруживается тельце Барра, а у мужчин — нет. В случае хромосомных аномалий телец Барра всегда на одно меньше, чем хромосом X.
Изучение строения и функционирования хромосому человека имеет большое теоретическое и практическое значение для медицинской генетики. Знание того, что представляет собой каждая хромосома человека в химическом, цитологическом и генетическом отношении, важно для правильного понимания происхождения хромосомных нарушений и обусловленных ими аномалий развития, а следовательно, и поиска путей исправления этих отклонений.
Хромосомные болезни клиницисты начали изучать еще до установления точного числа хромосом человека. Например, синдромы Клайнфелтера и Шерешевского — Тернера были четко описаны до открытия хромосомной этиологии этих заболеваний и хорошо известны врачам. К хромосомным болезням относят такие формы патологии, при которых наблюдаются, как правило, нарушения психики и множественные врожденные пороки различных систем организма человека. Генетической основой таких состояний являются хромосомные мутации — численные или структурные изменения хромосом, наблюдаемые в соматических или половых клетках.
Термин «болезнь» по отношению к хромосомным аномалиям, как аутосомных, так и половых хромосом, употребляется не совсем справедливо. Болезнь — это процессуальность, т. е. закономерная смена симптомов и синдромов во времени. Болезнь имеет продрому, начало, стадию полного развития и исходное состояние. Совокупность же специфических признаков, характеризующих любую хромосомную аномалию, является конституцио-нальной, врожденной и признаки эти непрогредиентны.
Большинство хромосомных болезней возникает спорадически в результате геномной и хромосомной мутаций в гаметах здоровых родителей или на первых делениях зиготы. Хромосомные изменения в гаметах приводят к развитию так называемых полных, или регулярных, форм нарушения кариотипа, а соответствующие изменения хромосом на ранних стадиях развития эмбриона являются причиной возникновения соматического мозаицизма, или мозаичных организмов (наличие в организме двух или более клеточных линий с разным числом хромосом). Мозаицизм может касаться как половых хромосом, так и аутосом. Мозаики, как правило, имеют более «стертые» формы заболевания, чем люди с измененным числом хромосом в каждой клетке.Так, ребенок с мозаичным вариантом болезни Дауна может иметь нормальный интеллект, но физические признаки этого заболевания остаются.
Число аномальных клеток может быть различным: чем их больше, тем более ярко выражен симптомокомплекс той или иной хромосомной болезни. В некоторых случаях удельный вес аномальных клеток так невелик, что человек кажется фенотипически здоровым.
В некоторых случаях установить мозаицизм оказывается не так просто, поскольку клон аномальных клеток имеет в онтогенезе тенденцию к элиминации. Иначе говоря, число таких клеток может быть у взрослого человека относительно мало, в то время как в эмбриональный и ранний постнатальный период их удельный вес был достаточно велик, что привело к развитию выраженных клинических симптомов болезни. Однако, несмотря на известные трудности изучения мозаицизма, его открытие и исследование вносят ясность в проблему стертых и рудиментарных форм хромосомных болезней.
В основе классификации хромосомных болезней лежат типы мутаций. Хромосомные мутации (числовые или структурные) возможны в соматических или половых клетках, они возникают в результате числовых или структурных изменений хромосом или их сочетания. Числовые изменения сводятся к наличию добавочных хромосом или отсутствию одной из хромосом. В первом случае говорят о трисомии по какой-либо из 23 хромосом, во втором — о моносомии. Реже можно наблюдать нарушение плоидности хромосомного набора (увеличение на полный гаплоидный набор).
Структурные изменения хромосом у человека хотя и встречаются намного реже, чем численные аберрации, представляют интерес как общетеоретический, так и клинический. Можно выделить два основных типа перестроек: внутрихромосомные и межхромосомные. В свою очередь перестройки могут быть сбалансированными, т. е. в геноме присутствуют все локусы, однако их расположение в хромосомах отличается от исходного нормального. Несбалансированные перестройки характеризуются утратой или удвоением участков хромосомы. Внутрихромосомные перестройки, связанные с перестройками внутри одного плеча хромосомы, называются парацентричес-кими. Крайние участки без центромеры называются фрагментами и они обычно утрачиваются в ходе митоза.
Деления — это утрата части хромосомы, происходящая в результате двух разрывов и одного воссоединения с утратой сегмента, лежащего между разрывами. У человека известна делеция хромосомы 5. Такая делеция выражается в синдроме «кошачьего крика». Дупликация—это удвоение сегмента хромосомы, в результате чего клетка организма становится полиплоидной по данному сегменту. Если дупликация находится непосредственно за исходным участком хромосомы, то это называется тандем-дупликацией. Кроме того, дупликации могут быть локализованы в других участках хромосомы. Большинство таких перестроек детальны, а те индивиды, которые с ними выжили, как правило, не способны оставить потомство.
В случае инверсии участок хромосомы разворачивается на 180° и разорванные концы соединяются в новом порядке. Если в инвертированный участок попадает центромера, то такую инверсию называют перицентрической. Если инверсия затрагивает только одно плечо хромосомы, то она называется парацентрической. Гены в инвертированном участке хромосомы располагаются в обратном по отношению к исходному в хромосоме порядке.
К межхромосомным перестройкам относят транслокации — обмен сегментами между хромосомами. Различают следующие типы транслокаций: 1) реципрокная транслокация, когда две хромосомы взаимно обмениваются сегментами; 2) нереципрокная транслокация, когда сегмент одной хромосомы переносится в другую; 3) транслокация типа центрического соединения, когда после разрывов в околоцентромерном районе соединяются два фрагмента с центромерами таким образом, что их центромера соединяется, образуя одну. Транслокационный синдром Дауна возникает именно таким образом. При этом больные имеют выраженную симптоматику болезни Дауна, но в их кариотипе всего 46 хромосом, причем хромосом 21 и Х — две, третья транслоцирована на хромосому группы D (возможно, хромосому 15). Исследование кариотипов их родителей показало, что чаще всего фенотипически нормальные матери имеют 45 хромосом и точно такую же транслокацию хромосомы 21, как и ребенок.
Хромосомные болезни можно классифицировать по тому, какая из систем хромосом — половая или аутосомная — вовлекается в патологический процесс. До настоящего времени точной общепринятой классификации хромосомных болезней нет. Это связано со многими причинами, в частности, с тем, что патогенетические механизмы хромосомных нарушений еще не выяснены. Большинство хромосомных аберраций по-прежнему относят к группе синдромов. Лишь некоторые из них можно назвать болезнями. Это в полной мере справедливо для болезней Дауна и Клайнфелтера.
Какова же общая клиническая характеристика хромосомных болезней? Почти все они сопровождаются множественными нарушениями скелета, психики. Отмечаются врожденные пороки наружных и внутренних половых органов, их замедленный рост. Нарушается деятельность нервной, эндокринной и других систем, снижена генеративная функция, наблюдается четкое повышение смертности среди лиц с хромосомными аномалиями.
Диагностические признаки разделяются на 3 группы. А — комплекс признаков, позволяющих лишь заподозрить хромосомную аномалию. Это общие признаки: физическое недоразвитие, ряд дизморфий мозгового и лицевого черепа (деформация ушных раковин и их низкое расположение, микроцефалия, эпикант, высокое небо), косолапость, клинодактилия мизинцев, некоторые пороки развития внутренних органов (сердца, почек, легких). В — признаки встречаются в основном при определенных хромосомных болезнях. Их сочетание позволяет в большинстве случаев диагностировать хромосомную аномалию. Среди характерных, наиболее часто встречающихся признаков этой группы при трисомии хромосомы 18 следует назвать долихоцефалию (89,6% случаев), флексорное положение кистей (96,1 %), «стопу-качалку» (76,2%), короткий и широкий I палец стопы (70,6% случаев); при трисомии по хромосоме 13—расщелину верхней губы и неба (68,7 % случаев), флексорное положение кистей (44,4%), косоглазие (31,4%), дефект скальпа (30,5 % случаев) и др. С — признаки характерны только для одной хромосомной аномалии, например, «кошачий крик»—при синдроме 5р—-, алопеция при синдроме 18р.
Хромосомным болезням свойственна чрезмерная фенотипическая (клиническая) вариабельность. Часто при одних и тех же хромосомных аномалиях клинические признаки выражены по-разному. В качестве примера можно привести болезнь Дауна, при которой поражение психики проявляется слабоумием от легких до тяжелых степеней (дебильность — имбецильность — идиотия). Выраженность клинических проявлений хромосомных болезней зависит от многих причин, среди которых следует отметить генотипические и паратипические факторы, состав поражаемых генов, размер аберрации и индивидуальность хромосомы, процент мозаичных клеток в организме и т. д. Иногда при низком содержании! мозаичных клеток клиническая картина бывает стертой." Это особенно часто наблюдается при мозаицизме по половым хромосомам. Обращает на себя внимание и то, что, как правило, клинические проявления у больных с аутосомными аберрациями намного тяжелее, чем у больных с нарушением в системе половых хромосом. Следовательно, жизнеспособность больных с аберрациями половых хромосом значительно выше. Среди новорожденных с хромосомными аберрациями около 50 % детей имеют аутосомные аномалии, а другие 50 % — аномалии по половым хромосомам, несмотря на то что система аутосом представлена 22 парами хромосом, а система половых хромосом — только одной парой.
Интеллект при аутосомных синдромах нарушается гораздо резче, чем при синдромах, вызванных аномалиями половых хромосом.
Клинические и цитогенетические исследования, проводимые у новорожденных с хромосомной патологией, показывают, что жизнеспособность их зависит от типа хромосомного нарушения. Большинство с аутосомными трисомиями погибают в первые дни жизни. У больных с аномалиями половых хромосом жизнеспособность, напротив, не снижена. Это связано с тем, что полная клиническая картина у больных данного контингента разворачивается лишь в период полового созревания, когда начинают функционировать гены, определяющие половое развитие организма и формирование вторичных половых признаков. Из других контингентов хромосомные- аномалии обнаруживаются: среди детей с олигофренией в среднем у 15 % больных (в основном структурные перестройки); у больных с нарушением половой дифференцировки частота хромосомных нарушений колеблется от 20 до 50 % ( у 50 % из них обнаруживается мозаицизм); у больных с первичной и вторичной аменореей частота хромосомных аномалий колеблется от 10 до 50 % (более 90 % — численные нарушения и мозаицизм); при мужском бесплодии частота аномальных хромосом достигает 10—15% (до 70 %—численные нарушения и мозаицизм).При отягощенном акушерском анамнезе у супружеских пар с повторными спонтанными абортами, мертворождениями или рождением детей с пороками развития сбалансированные перестройки наблюдаются в 5 % случаев.
Поделиться316.08.2011 15:16
Для диагностики хромосомных болезней в настоящее время применяют ряд методов медицинской генетики, чаще клинико-генеалогический, цитогенетический (определение полового хроматина и кариотипирование), пато-логоанатомический и дерматоглифический. Некоторые хромосомные болезни можно диагностировать клинически, не прибегая к другим методам. Например, своеобразие клиники синдромов Шерешевского —Тернера и Клайнфелтлера позвлояет опытному клиницисту поставить диагноз без цитогенетического анализа.
Как правило, современная диагностика любого заболевания является комплексной. Кроме традиционных клинических данных, лабораторных исследований, сбора анемнестических данных,, при диагностике наследственных болезней, в частности хромосомных, особое внимание уделяется изучению гениалогии больного. Только около 3-5% их четко наследуется.
Основным методом диагносики хромосомных болезней является цитогенетический, который включает в себя: а) определение полового хроматина; б) определение "барабанных палочек"; в) определение добавочной хромосомы Y с помощью флюоресцентной микроскопии; г) кариоптирование (получение хромосомных наборов). Наиболее точным и достоверным методом исследований является кариологический.
Из вспомогательных методов диагностики хромосомных заболеваний наиболее прост и доступен дерматоглифический метод, применяемый для анализа кожных узоров на ладонях, подошвах и сгибательной поверхности пальцев, так как при хромосомных болезнях наблюдается специфическое изменение кожных узоров.
Основными показаниями для направления на цитогенетическое обследование больного и его родственников являются: 1) наличие лиц с выявленной паталогией полового хроматина; 2) наличие детей с множественными пороками развития; 3) олигофрения в сочетании с чертами внутриутробного дисгенеза или врожденными пороками развития; 4) повторные спонтанные аборты у женщин, мертворожденные дети в анамнезе или дети с пороками развития (обследованию подлежат и мужья); 5) наличие в анамнезе умерших детей с множественными врожденными пороками развития или установленным хромосомным синдромом; 6) наличие структурной перестройки и сбалансированного носительства транслокации или инверсии у матери или отца пробанда; 7) необходимость определения кариотипа плода у женщин с высоким риском рождения ребенка с хромосомной паталогией.
2. Стоматологические проявления наследственных болезней и синдромов.
2.1 Генетические аспекты кариеса, болезней пародонта,расщелены верхней губы и неба.
Известно, что на резистентность зубов к кариесу влияют многие генетические факторы. А. А. Зубов и Л. Т. Левченко (1981), И. А. Бальчюнене (1985) отмечали, что в известной степени резистентность зависит от морфологических признаков зубов (наличие и строение бороздок, ямок, размеры зуба, его дифференцированность). У лиц, резистентных к кариесу, более архаичное строение жевательной поверхности первого верхнего моляра, а у больных с множественным кариесом сильнее выражены эволюционно сравнительно «молодые» вариабельные особенности строения верхних моляров. М. Brucker (1944) также подчеркивал влияние наследственности на конституцию зуба и его предрасположенность к кариесу.
М. Л. Гликман (1977) отмечал популяционное многообразие клинической картины кариеса, четко выраженные индивидуальные количественные характеристики процесса. Наследственность влияет как на резистентность зубов к кариесу, так и на их предрасположенность к кариозному процессу, выраженность которого генетически детерминирована.
Г. Н. Пахомов и соавт. (1979), изучая вопрос о связи кариеса зубов и факторов наследственности, выдвинули гипотезу о влиянии наследственности на резистентность зубов к нему. Неполная пенетрантность и высокий коэффициент наследуемое™ дают основание предполагать, что в генетическую систему, детерминирующую резистентность зубов к кариесу, вовлечены более чем один ген. Определенный ген (главный) контролирует возможность развития данного признака. Он рецессивен по отношению к гену, детерминирующему предрасположенность к карие-
Резистентность зубов к кариесу определяется не только морфологическими признаками, но и состоянием иммунной системы. А. И. Рыбаков и В. С. Иванов (1980) указывали на наследственную предрасположенность к кариесу, которая может проявляться уже в период закладки и развития органа, а также зависит от состояния иммуниой системы организма.
В последние годы изучение наследственной предрасположенности к кариесу зубов ведется в направлении исследования ассоциации генетических маркеров крови (эритроцитарные антигены системы АВО, MN, Rhesus и др.) с интенсивностью кариеса. М. Л. Гликман (1973), Ф. 3. Савранский и соавт. (1985), М. Н. Травицкая и соавт. (1985), А. И. Марченко и соавт. (1984, 1986), В. Gawrzewska (1978) установили ассоциативную связь между интенсивностью кариеса зубов и рядом эритроцитарных антигенов. Интенсивный кариозный процесс одинаково часто наблюдается у больных с различными группами крови. По-видимому, гемагглютинины крови, для которых установлена связь с интенсивностью кариеса зубов, самостоятельного значения в его развитии не имеют, а действуют синергично с другими защитно-приспособительными механизмами гомеостаза.
В настоящее время общепризнана мультифакториальная этиология кариеса зубов в нормальной популяции. Резистентность зубов к нему во многом зависит от их морфологии, а также от иммунного статуса. Слюнные железы вырабатывают им-муноглобулины класса A (IgA), продукция которых не зависит от содержания их в сыворотке крови [Рыбакова М. Г., 1979, и др.]. Колебания уровня IgA и других иммуноглобулинов в слюне человека являются важным фактором, определяющим возможность возникновения и развития патологического процесса в полости рта. Следовательно, рези стентн ость зубов к кариесу также ассоциирована с вариабельностью паротидной слюны и составом белков ротовой жидкости.
Наследственная форма фиброматоза десен сравнительно редкое стоматологическое заболевание, которое может проявиться на первом году жизни или на десятом году жизни ребенка. Чаще фиброзные разрастания ограничиваются определенной группой зубов, но возможен генерализованный фиброматоз десен; коронки зубов закидываются на 2/3 или полностью. Наследственные формы фиброзно- гиперпластических изменений слизистой оболочки рта обычно имеют аутосомно- доминантное наследование. Редко бывают рецессивные формы с различной пенетрантностью. Возможны спорадические случаи. Симметричные фибромы полости рта также имеют аутосомно- доминантное наследование, но возможны рецессивные формы с низкой пенетрантностью. В.С. Иванов (1981) относит фиброматоз десен к генетически обусловленным заболеваниям. Лечение фиброматоза десен хирургическое: если патологический процесс расположен у временных зубов, они подлежат удалению.
В настоящее время известно более 50 синдромов, которые в качестве одного из признаков включают расщелину губы и/ или неба: у 1% больных с типичной расщелиной губы, челюсти и неба наблюдается аномалия развития уха, популяционная частота выраженной аномалии развития уха равна 0,07% ; у 15,4% больных констатируют генетическую основу расщелины губы и неба; у 19,57 % больных расщелина носила семейный характер; у 22,9 % больных установили наследственную причину расщелины губы, челюсти и неба, степень наследования двусторонней расщелины губы, челюсти и неба более высока (30,6 %).; у 3,3 % больных с врожденным пороком развития лица обычно выявляют феноскопии (угрожающий выкидыш в I триместре беременности, лучевое и гормональное воздействие, тяжелая форма раннего токсикоза беременных ); у 17 % больных с расщелиной губы, челюсти и неба выявил ее семейный характер.
Анализ дерматоглифических отпечатков пальцев и ладоней показал, что при врожденной расщелине губы и неба они аномальны по сравнению с контролем. Дерматоглифические исследования R.N.Deshmukh и соавторы (1979), N. Kanematsu (1982) для выяснения этиологии этой аномалии развития показали, что при семейных случаях расщелины губы и неба несимметричны отпечатки правой и левой руки, а в тератогенной и в контрольной группе они симметричны. Автор считает, что этот порок развития мультифакториален.
В заключение следует отметить, что еще не ясны генетические и биохимические механизмы, влияющие на возникновение расщелин губы и неба у человека. Тератологические исследования, которые проводят в настоящее время, помогутуглубить знания об этиологии расщелины и выработать меры профилактики. В первые 3 мес. беременности женщины должны исключить прием любых лекарственных средств, включая домашние.
По данным M.M. Cohen (1976), более 100 синдромов могут сочетаться с различными расщелинами лица: из них 30 синдромов наследуются по аутосомно- доминантному типу, 35- по аутосомно- рецессивному типу, 6 сцеплены с плодом, 22 обусловлены хромосомными аберрациями и 7 имеют неясный тип наследования.
2.2 Наследственные заболевания твердых тканей зубов.
Довольно большая группа заболеваний зубов связана с поражением эмали. Этиологическим фактором наследственных заболеваний эмали являются патологические мутантные гены, которые передаются больному через половые клетки родителей. Несовершенный амелогенез , дисплазия эмали, группа наследственных дефектов, характеризующихся нарушением обмена веществ на одном из двух этапов образования эмали: неправильное формирование матриц ведет к гипоплазии эмали, а нарушение созревания- к ее гипокальцификации. Изменения эмали могут быть обусловлены двумя причинами: генной мутацией и факторами окружающей среды или их сочетанием. Нарушение процессов формирования матрикса эмали ведет к полному, частичному или локальному изменению ее толщины, что выражается в ряде клинических форм наследственной гипоплазии эмали.
Нарушение созревания эмали, связанное с изменением обызвествления матрицы, вызывает целый ряд клинических и морфологических дефектов: дезорганизацию эмалевых призм, крайне низкую степень кристализации, одиночные неравномерно расположенные кристаллы гидрооксиапатита, изменение пластичности, окраски и толщины эмали. Из всех наследственных заболеваний эмали наиболее распространена гипоплазия, связанная с недостаточным обызвествлением эмалевых призм. При этом органическое содержание эмали возрастает до 8,7- 14,2%.
Следовательно, все наследственные заболевания эмали по клинико- морфологическим признакам могут быть разделены на 3 основные группы, каждая из которых имеет клинические разновидности:
Наследственная гипоплазия эмали, связанная с нарушением ее матрикса:
аутосомно- доминантная точечная гипоплазия,
аутосомно- доминантная локальная гипоплазия,
аутосомно- доминантная гладкая гипоплазия,
аутосомно- доминантная шероховатая гипоплазия,
аутосомно- рецессивная шероховатая аплазия эмали,
сцепленная с Х- хромосомой доминантная гладкая гипоплазия.
Наследственная гипоплазия эмали, связанная с нарушением ее созревания.
аутосомно- доминантное гипосозревание в сочетании с тавродонтизмом,
сцепленное с Х- хромосомой рецессивное наследование, гипосозревание,
аутосомно- рецессивная пигментация, гипосозревание,
"снежная шапка"- аутосомно- доминантное гипосозревание.
Наследственная гипоплазия эмали, связанная с ее гипокальцификацией (гипокальцинозом):
аутосомно- доминантная гипокальцификация,
аутосомно- рецессивная гипокальцификация.
2.3. Генетически обусловленные аномалии зубов.
Размеры и форма зубов в известной степени зависят от системы генов, ответственных за рост и развитие, которые находятся в комплексе половых хромосом XY. Так L. Alvesalo и A. Chapelle (1979), М. Kari и соавт. (1980), L. Alvesalo и Р. Portin (1980), Р. Kir-veskari и L. Alvesalo (1981), G. Klein и D. Eismann (1986) установили изменение размеров зубов при аномалии комплекса половых хромосом. У девочек при кариотипе 45.ХО (синдром Шерешевского — Тернера) уменьшены размеры временных зубов, у мужчин с 47XXY увеличены коронки постоянных. Изменения размеров зубов были выявлены при других аномалиях комплекса половых хромосом при синдроме мужчин 46ХХ, у мужчин с кариотипом 47XYY, с делецией длинного плеча Y-хромосомы. Следовательно, изменения размеров зубов вызваны генетическим эффектом и подтверждают концепцию существования специфического гена роста в Х и Y хромосомах человека.
Время прорезывания, скорость обызвествления временных и постоянных зубов ряд авторов рассматривают как семейный признак, т. е. эти процессы связывают с генетическими факторами [Garn S. М. et al., 1965; Bartosova A., 1973]. Популяционное исследование цвета зубов Е. Райчиновой (1976) показало, что цвет зубов является наследственным признаком.
В настоящее время форма передачи гиподонтии изучена в достаточной степени. Установлено влияние генетических факторов на развитие адентии. Врожденное отсутствие зубов—наследственное явление, которое может проявляться в каждом поколении, ( чаще — у родственников первой степени родства).
В настоящее время установлена наследственная этиология таких распространенных аномалей, как шизодонтия, инвагинация зуба, сверхкомплектные зубы.
Шизодонтия. Аномалийные «двойные» зубы (шизодонты) образуются в результате расщепления зачатка зуба, чаще верхних резцов, реже нижних, и крайне редко премоляров. Различают 3 формы этой аномалии: расширение коронки; вертикальное, губное и оральное ее расщепление — близнецовые зубы; разделение на два самостоятельных сросшихся зуба — синодонтия.
Инвагинация (dens in dente). Одонтома внутри зуба—это редкая аномалия развития зубов.Эта аномалия обычно возникает в ранний период развития эпителиальной диафрагмы.
2.4. Изменения зубочелюстной системы при хромосомных болезнях.
Хромосомными болезнями называют группу заболеваний, вызванных числовыми или структурными аберрациями хромосом, видимыми в световой микроскоп. Примерно 1 % всех новорожденных имеют хромосомные аномалии, ведущие к серьезным последствиям. Приблизительно 90 % этих аномалий приходится на долю анеуплоидий, половину всех случаев составляет аутосомные анеуплоидий, а половину — анеуплоидий по половым хромосомам. При некоторых типах анеуплоидий бывает затронута лишь часть клеточных линий, что приводит к мозаицизму. Другие хромосомные аномалии относятся к категории структурных и представляют собой утрату участков хромосом, перестройку хромосомного материала, транслокации и др. [Стивенсон А., Дэвисон Б., 1972].
По данным Г. И. Лазюка, И. В. Лурье (1979), )'S. Denes (1982), анализ основных проявлений почти '25 хромосомных заболеваний, обусловленных аберрациями аутосом, показал, что у одного ребенка , удается установить не менее 20 врожденных анома-. лий и пороков развития. Челюстно-лицевые изме-. нения не всегда являются обязательными признаками и очень разнообразны: деформация черепа, брахицефалия, плоское лицо, гипоплазия средней его части, запавшее переносье, выступающее надпереносье; гипертелоризм, косоглазие, монголоидный j разрез глазных щелей, эпикантус; густые широкие | брови, синофриз, низко расположенные и деформи-: рованные ушные раковины; клювовидный нос; микрогения, макростомия, «рыбий» рот, расщелина неба, 1 выступающий изо рта складчатый язык.
Хромосомы 5р, или «кошачьего крика» (cri du chs синдром, связан с делецией 5-й хромосомы группы В.|
По данным Г. И. Лазюка, И. В. Лурье, для него характерно сочетание черепно-лицевой дисплазии характерным плачем, напоминающим кошачье мяуканье. Эта болезнь имеет следующие признаки: круглое асимметричное лицо (66%), микроцефалия (91%), седы волосы, широкое запавшее переносье, наружные верстия носа сужены, углы рта опущены, микрогния (79 %), гипертелоризм, антимонголоидный разрез глазных щелей, косоглазие, эпикантус, низко распо-ложенные ушные раковины, иногда деформированны (козелок и противозавиток не выражены). У 305 больных имеется аномалия органа зрения (атрофия зрительных нервов и др.). Наиболее постоянный признак синдрома — «кошачий крик» — связан с аномалией гортани (уменьшение надгортанника и др.) Следует отметить, что такие диагностические признаки, как «кошачий крик», мышечная гипотонии круглое лицо, у большинства больных с возрастом исчезают. У 11 % больных отмечают синдактилию стоп, а также клинодактилию V пальца кисти. Пороки сердца выявляют у 15—30 % больных. Характерно умственное и физическое недоразвитие. Половые органы сформированы правильно, но у мальчиков бывает крипторхизм (15%). Продолжительность жизни не установлена.
Хромосомы 13 трисомии, или Патау синдром, как правило, вызывается присутствием лишней хромосомы в группе D (13—15). Внешние признаки синдрома Патау настолько типичны, что позволяют диагностировать его при осмотре. Особенно характерны аномалии черепа и лица: микроцефалия в сочетании с низким и скошенным лбом, узкие глазные щели, гипотелоризм, запавшее переносье и широкое основание носа, низко расположенные и деформированные ушные раковины с гипоплазированным козелком, дефект скальпа теменной области /30%). Расщелина верхней губы и неба встречается у 68 % больных, в том числе только неба—у 10%. Часто бывает двусторонняя расщелина губы. Типичны микрофтальмия (70%), эпикантус, колобома радужки, дисплазия сетчатки и другие аномалии глаз.
Возможны также капиллярная гемангиома лба и затылочной области, отсутствие бровей, подглазничный край не выражен. Недоразвиты обе челюсти, расщеплен язык .
У 49 % больных наблюдаются полидактилия кистей, реже — стоп, а также флексорное положение пальцев рук. Часты пороки внутренних органов: ги-поплазия мозолистого тела, мозжечка, врожденные пороки сердца (80%), нарушение развития поджелудочной железы, аномалии почек. Всегда поражены половые органы (крипторхизм, двурогая матка и др.).
Дети с синдромом Патау живут недолго; по данным литературы, 90 % умирают в возрасте до 1 года. Все выжившие дети слабоумны. Операция по поводу расщелины губы, неба нецелесообразна, поскольку больные дети обычно нежизнеспособны.
Популяционная частота от 1:5000 до 1:7800.
Хромосомы 18 трисомии, или Эдвардса синдром, так же как и предыдущий, связан с наличием добавочной хромосомы в группе Е (18).
Для него характерны следующие клинические признаки: мозговой череп долихоцефалической формы со ступенеобразным западением лобных костей в области родничка, узкие глазные щели, надпереносье слегка выступает. Ушные раковины деформированы и в большинстве случаев низко расположены, могут отсутствовать мочки, козелок. Отмечают также гипоплазию нижней челюсти, микростомию, у 15 % больных — расщелину неба, короткую верхнюю губу. Вероятны, кроме того, микроцефалия (40%), выступание затылка, выдающиеся скулы, гирсутизм лба, микрофтальм, птоз век, эпикантус, колобома радужки. Возможны ограничение открывания рта, атрезия хоан (10%). Этот синдром чаще наблюдается у девочек. Популяционная частота 1:7000.
Хромосомы 21 трисомии, или Дауна синдром, или монголизм, является наиболее распространенной хромосомной патологией у человека. Основные клинические признаки заболевания следующие: округлая форма головы, скошенный и узкий лоб, плоское лицо, короткий нос, плоская переносица, монголоидный разрез глаз, эпикантус, возможен избыток кожи на шее, а также диспластичные уши, толстые губы, увеличенный, часто «складчатый» язык.
Из внутренних органов чаще всего поражается сердце (60%), возможны атрезия или стеноз двенадцатиперстной кишки (6%), аномалии почек (6 %). У 45 % больных возможно поставить предварительй диагноз на основании дерматоглифики (четырехпальцевая сгибательная складка, одна сгибательная складка на V пальце), у 60 % есть клинодактия V пальца. Для 90 % детей с синдромом Дауна характерна глубокая умственная отсталость.
Стоматологические аспекты синдро Дауна. У 93 % больных задерживается проре-вание временных зубов; описаны случаи, когда их прорезывание заканчивалось лишь к 4 годам. Задержка прорезывания постоянных зубов наблюдается у 64 % больных.
Частота кариеса временных зубов при синдроме Дауна ыше по сравнению с показателем у практически здоровых детей. У больных нередко имеется гипоплазия моляров. Меньшую частоту кариеса постоянных зубов больных с синдромом Дауна, по-видимому, можно объяснить морфологической аномалией коронок зубов:
архаичные, предковые черты у премоляров и вторых ноляров верхней челюсти. Статистический анализ гипсовых моделей зубов показал, что у 30 % больных имелась аномалия их коронок: первого премоляра нижней челюсти (72%), второго премоляра (59%) и второго моляра (61 %) верхней челюсти.
Известно, что одним из признаков этого синдрома является тяжелая форма пародонтита. Динамическая ортопантомография выявила пародонтит у 69—75 % больных и у 20^43 % членов контрольной группы (умственно отсталые больные из специализированного учреждения). У больных с синдромом Дауна моложе 30 лет, очень часты быстро прогрессирующие изменения пародонта в области передних зубов и первых моляров. Они обусловлены следующими факторами: аномалией капилляров пародонта, изменением соединительной ткани, нарушением активности полиморфно-ядерных лейкоцитов, моноцитов и уменьшением количества Т-клеточных лимфоцитов. У больных с синдромом Дауна, находящихся в специальных учреждениях, изменения в пародонте более выражены, чем у живущих дома. Объясняется это стрессом, что присуще этим учреждениям. Для синдрома характерен тяжелый пародонтит с горизонтальной резорбцией межзубных перегородок кости, особенно выраженной в области передних зубов нижней челюсти.
Хромосомы Х моносомии, или Шерешевского-Тернера синдром, или ХО-синдром,— аномалия полового развития у женщин, выражающаяся в нарушении половой дифференциации: половые железы представляют собой соединительнотканные тяжили рудименты яичника. Синдром имеет четыре основ- ных признака: низкий рост, инфантилизм, недоразвитие гонад и сопутствующие соматические аномалии (у 25 % больных пороки сердца, аномалии почек и др.). Чаще всего типичные клинические проявления синдрома связывают с кариотипом 45,XO или мозаицизмом 45,ХО/46,ХХ. В связи с этим важным диагностическим тестом является исследование полового хроматина в ядрах клеток буккального эпителия, которое указывает на отсутствие или аномалию одной из половых хромосом. Изменена дерматоглифика — поперечная ладонная складка и др.
Признаком заболевания у новорожденных служат лимфатический отек тыльных поверхностей стоп, голени, кистей, шеи, а также врожденные крыловид-ные кожные складки на шее (32%). Изменения скелета очень разнообразны, нарушено как энхондраль-ное, так и периостальное костеобразование.
Лицо больных нередко имеет старческие черты, углы рта опущены. Как частый симптом можно также отметить миндалевидные глаза, подчеркнутые гиперпигментацией век или наружных углов глаз. Отмечают птоз век, эпикантус, косоглазие, астигматизм, голубые склеры.
Заключение.
Стоматолог может внести определенный вклад в раннюю диагностику многих наследственных болезней и синдромов. Такие заболевания, как полипоз кишечника, тип II (синдром Пейтца- Егерса), полипоз кишечника, тип III (синдром Гарднера), гистиоцитоз X, синдром базально- клеточного невуса ( синдром Горлина) и синдром Ван- дер- Вуда, а атк же гиперпаратиреоз первичный (семейный), врожденная аплазия щитовидной железы и другие, впервые может обнаружить стоматолог и после консультации с хирургом, эндокринологом и другими специалистами поставить правильный диагноз.
Для диагностики наследственных заболеваний особое значение имеет осмотр лица: разрез глазных щелей (монголоидный или антимонголоидный), птоз, эпикантус, экзофтальм, помутнение хрусталика, голубые склеры, косоглазие, размер ресниц, преносица (запавшая, широкая), выступающие надбровные дуги, аномалии фильтра и красной каймы верхней губы, гипо- и гипертелоризм, носолобный угол, пигментация кожи лица слизистой оболочки полости рта, а также форма и положение ушной раковины, форма черепа, позднее закрытие родничков, имеют значение рост, положение пальцев кистей, анализ дерматоглифики, умственная отсталость и другие признаки.
Известно, что этиологическим фактором наследственных заболеваний являются патологические мутантные гены, которые передаются больному через половые клетки родителей. Для предупреждения появления в семье больных с наследственной патологией большое значение имеет медико- генетическое консультирование, основными задачами которого являются: установление точного диагноза заболевания, определение тиа его наследования в данной семье, расчет величины риска повторения, объяснение смысла медико- генетического прогноза обратившимся.
Список литературы.
Беляков Ю.А. Стоматологические проявления наследственных болезней и синдромов.- М.: Медицина,1993
Лильин Е.Т., Богомазов Е.А., Гоман- Кадошников П.Б. Генетика для врачей- М., Медицина, 1990
Колесов А.А., Каспарова Н.Н., Жилина В.В. Стоматология детского возраста- М.: Медицина,1991
Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. Наследственные синдромы и медико- генетическое консультирование.Справочник.-Л.:Медицина,1989
Лазюк Г.И., Лурье И.В., Черствой Е.Д. Наследственные синдромы множественных пороков развития.-М.:Медицина,1983
Наследственные болезни: Справочник/ Под ред. Л.О. Бадаляна.- Ташкент:Медицина,1980
Опубликовано: 2005-05-23
http://www.medicreferat.com.ru/pageid-1096-5.html
Поделиться416.08.2011 15:22
медицинский форум..здесь можно задать свои вопросы и получить ответы от врачей!
http://forums.rusmedserv.com/forumdispl … amp;page=2
Поделиться516.08.2011 15:24
Клинические аномалии в системе аутосом
Возможные исходы беременности при сегрегации продуктов сбалансированных носителей робертсоновской транслокации.
«Обычная» трисомия включает в себя 95% всех случаев синдрома Дауна. Пример, но в 1% случаев она обусловлена мозаицизмом (это, несомненно, минимальное количество, так как определенное число лиц с мозаицизмом, возможно, остаются невыявленными, особенно у фенотипически здоровых родителей, у которых родились дети с трисомией); в остальных случаях трисомия обусловлена транслокацией хромосом.
Основная часть транслокаций, обусловливающих синдром Дауна, относится к центральному соединению 21-й и D-хромосом; примерно половина из них наследуется. Основное количество составляют (14q21q), редко встречаются t(15q21q).
Редкие случаи t(13q21q), возможно, объясняются отсутствием синдрома трисомии 13 у потомков фенотипически здоровых носителей транслокации Dq21q.
От женщин-носителей рождаются дети трех типов: с нормальным фено- и кариотипом, фенотипически здоровые носители и с транс локационной трисомией 21. Теоретически все три типа потомков должны встречаться с одинаковой частотой, но только у 10% из них отмечалась патология, возможно, из-за повышенной гибели несбалансирован ной зиготы или плода.
Частичное распределение, при котором видна небольшая Ph-хромосома (№ 22), сформированная в результате транслокации дистальной части длинного плеча хромосомы 22 на длинное плечо хромосомы 9; определяется в форме диска с яркой флюоресценцией.
Ожидаемая частота поражения наблюдалась у 1/3 плодов в ранние сроки беременности. От мужчин-носителей редко рождаются больные дети, чаще от них рождаются здоровые дети или носители. Только в 5% случаев транслокационная форма синдрома Дауна, в том числе две G-хромосомы, наследуются от родителя-носителя. Незначительную метацентрическую транслокацию хромосомы может представлять слияние хромосом 21 и 22 или двух хромосом 21 или неправильное деление центромеры с образованием изо-хромосомы 21.
Низкая частота наследования позволяет предположить, что большинство из этих метацентрических хромосом относится к последним двум типам, поскольку у здорового носителя может отмечаться редкое совпадение транслокации у одного из родителей в сочетании с отсутствием или потерей хромосомы 21 в гамете другого или вследствие транслокации после стадии зиготы.
У всех жизнеспособных потомков, рожденных от носителя t(21q21q), будет синдром Дауна (за исключением, возможно, лиц с моносомией 21). У носителей t(21q22q), с другой стороны, может родиться как носитель или здоровый ребенок, так и больной. Не все транслокации, обусловливающие синдром Дауна, относятся к типу центрического слияния. Некоторые исследователи сообщают об увеличении длинного плеча одной хромосомы 21.
«Болезни плода и новорожденного», проф. И.М. Воронцов
Поделиться616.08.2011 15:37
О предимплантационной диагностике в Германии
Политики против науки, поотрывала бы им руки,
Поотрывала бы им ноги, настолько мысли их убоги,
В погоне за "постом на блюде" не думают они о людях,
Иль мыслят лишь о людях "вместе", себя не ставя на их место,
В Германии идут дебаты: "Рожайте ТО, чем вы богаты,
Пусть будет у ребёнка хвостик, не надо РАННИХ ДИАГНОСТИК!
Вскрывайте лучше себе вены, раз не в порядке ваши гены,
Больных рожайте и убогих, мы не поможем, мы - не боги!"
И Меркель вспомню "добрым" словом, она сама жива-здорова,
И ей самой, и её мужу ген-тест, конечно, был не нужен,
Легко давать советы эти, когда здоровы дома дети,
Когда у сына и у дочки здоровы лёгкие и почки,
Но прежде чем лишать надежды, примерьте эти вот одежды:
С одного сайта:
"Не знаю насколько актуальна эта тема для других, но я столкнулась с этой проблемой и ищу способы как ее решить правильно.
Немного о себе:
Первый ребенок - мальчик - умер через 1 час после родов. Перед родами поставили диагноз: аутосомно-рецессивный поликистоз почек. На вопрос откуда это заболевание, все отвечали по разному: экология, случайность, питание, генетика.
На консультации у генетиков, они конечно все подтвердили, что это генетическое заболевание. Для анализов, чтобы узнать какие гены у нас с мужем мутируют, что дают наследственность детям именно это заболевание, в первую очередь нужен материал больного. Чего у нас конечно не было. Анализы соответственно не делали, генетики посоветовали не выбрасывать деньги.
Пока готовились к ЭКО, забеременили самостоятельно. Радости не было предела. Забыли обо всех болезнях и думали, что у нас все будет хорошо: "дважды снаряд в одну воронку не падает". Но..... на 27 неделе на очередном УЗИ прозвучал опять страшный диагноз: аутосомно-рецессивный поликистоз почек. Предложили прервать беременность, но я отказалась, решила бороться до последнего. Родила на 38 неделе. У ребенка почки были увеличены в несколько раз и занимали всю брюшную полость, соответственно сместив все остальные органы. Я не говорю уже о гипоплазии печени, надпочечников, легких (т.е. недоразвитость этих органов). Сын умер на 5 сутках. Паталоганат подтвердил диагноз и сказал, что это только генетическое заболевание. Врач Г в роддоме сказала, что скорей всего у нас это идет по мужской линии, т.е. порекомендовала рожать девочек, у которых возможно этого заболевания не будет.
Поделиться705.09.2011 20:22
- солнечная мама

- Зарегистрирован: 05.07.2011
- Приглашений: 0
- Сообщений: 1373
- Уважение: +17
- Провел на форуме:
18 дней 17 часов - Последний визит:
14.11.2011 12:20
Сейчас существует исследоание под названием CGH ( сравнительная
геномная гибридизация), когда проверяются все гены данного человека. При этом выявляются малейшие изменения в геноме.
Исследование это сравнительно недавнее и достаточно дорогое. У нас оно ещё не входит в корзину страховки больничных касс и стоит, примерно, 1200-1500 &. Интерпретировать этот анализ должен очень
грамотный специалист, прошедший соответствующую подготовку.
В России, как я полагаю, это исследование делается в Томске в ин-те
Медгенетики.
Очень надеюсь, что у Вас всё будет хорошо.
Поделиться816.10.2011 18:14
Классификация мутаций
1. По способу возникновения. Различают спонтанные и индуцированные мутации
Спонтанные происходят в природе крайне редко с частотой 1-100 на
миллион экземпляров данного гена. В настоящие время очевидно, что
спонтанный мутационный процесс зависит как от внутренних, так и от
внешних факторов, которые называют мутационным давлением среды.
Индуцированные мутации возникают при воздействии на человека
мутагенами –факторами, вызывающими мутации. Мутагены же бывают трех
видов:
. Физические (радиация, электро – магнитное излучение, давление,
температура и т.д.)
. Химические (цитостатики, спирты,фенолы и т.д.)
. Биологические (бактерии и вирусы )
2. По отношению к зачатковому пути. Существуют соматические и генеративные
мутации. Генеративные мутации возникают в репродуктивных тканях и
поэтому не всегда выявляются. Для того, чтобы выявилась генеративная
мутация, необходимо, чтобы мутантная гамета учавствовала в
оплодотворении.
3. По адаптивному занчению. Выделяют положительные, отрицательные и
нейтральные мутации. Эта классификация связана с оценкой
жизнеспособности образовавшегося мутанта.
4. По изменению генотипа. Мутации бывают генные, хромосомные и геномные
геномные.
5. По локализации в клетке. Мутации делятся на ядерные и
цитоплазматические. Плазматические мутации возникают в результате
мутаций в плазмогенах, находящихяс в митохондриях. Полагают, что именно
они приводят к мужскому бесплодию. Причем такие мутации в основном
наследуются по женской линии.
Генные мутации
Генные ( точковые ) мутации затрагивают, как правило, один или
несколько нуклеотидов, при этом один нуклеотид может превратиться в другой,
может выпасть(делеция), продублироваться, а группа нуклеотидов может
развернутся на 180 градусов. Например, широко известен ген человека,
ответственный за серповидно – клеточную анемию, который может привести к
летальному исходу. Соответствующий нормальный ген кодирует одну из
полипептидныз цепей гемоглобина. У мутантного гена нарушен всего один
нуклеотид (ГАА на ГУА). В результате в цепи гемоглобина одна аминокислота
заменена на другую( вместо глутамина – валин). Казалось бы ничтожное
изменение, но оно влечет за собой роковые последствия: эритроцит
деформируется, приобретая серповидно – клеточную форму, и уже не способен
транспортировать кислород, что и приводит к гибели организма. Генные
мутации приводят к изменению аминокислотной последовательности белка.
Наиболее вероятное мутация генов происходит при спаривание тесно связанных
организмов, которые унаследовали мутантный ген у общего предка. По этой
причине вероятность возникновения мутации повышается у детей, чьи родители
являются родственниками. Генные мутации приводят к таким заболеваниям, как
амавротическая идиотия, альбинизм, дальтонизм и др.
Интересно, что значимость нуклеотидных мутаций внутри кодона
неравнозначна: замена первого и второго нуклеотида всегда приводит к
изменению аминокислоты, третий же обычно не приводит к замене белка. К
примеру, "Молчащая мутация"- изменение нуклеотидной
последовательности, которая приводит к образованию схожего кодона, в
результате аминокилотная последовательность белка не меняется.
Хромосомные мутации
Хромосомные мутации приводят к изменению числа, размеров и
организации хромосом, поэтому их иногда называют хромосомными
перестройками. Хромосомные перестройки делятся на внутри- и межхромосомные.
К внутрихромосмным относятся:
. Дубликация – один из участков хромосомы представлен более одного
раза.
. Делеция – утрачивается внутренний участок хромосомы.
. Инверсия –повороты участка хромосомы на 180 градусов.
Межхромосомные перестройки (их еще называют транслокации) делятся на:
. Реципрокные – обмен участками негомологичных хромосом.
. Нереципрокные – изменение положения участка хромосомы.
. Дицентрические – слияние фрагментов негомологичных хромосом.
. Центрические – слияние центромер негомологичных хромосом.
Хромосомные мутации проявляются у 1% новорожденных. Однако интересно,
исследования показали, что нестабильность соматических клеток здоровых
доноров не исключение, а норма. В связи с этим была высказана гипотеза о
том, что нестабильность соматических клеток следует рассматривать не только
как патологическое состояние, но и как адаптивную реакцию организма на
измененные условия внутренней среды. Хромосомные мутации могут обладать
фенотипическими явлениями. Наиболее распостраненный пример - синдром
"Кошачьего крика" (плачь ребенка напоминает мяукание кошки). Обычно
носители такой делеции погибают в младенчестве. Хромосомные мутации часто
приводят к паталогическим нарушениям в организме, но в то же время
хромосомные перестройки сыграли одну из ведущих ролей в эволюции. Так, у
человека 23 пары хромосом, а у обезьяны - 24. Таким образом различие
составляет всего одна хромосома. Ученые предполагают, что в процессе
эволюции произошла хотя бы одна перестройка. Подтверждением этого может
служить и тот факт, что 17 хромосома человека отличается от такой же
хромосомы шимпанзе лишь одной перецентрической инверсией. Такие рассуждения
во многом подтверждают теорию Дарвина.
Геномные мутации
Главная отличительная черта геномных мутаций связана с нарушением
числа хромосом в кариотипе. Эти мутации так же подразделяются на два вида:
полиплоидные анеуплоидные.
Полиплоидные мутации ведут к изменению хромосом в кариотипе, которое
кратно гаплоидному набору хромосом. Этот синдром впервые был лишь обнаружен
в 60-ых годах. Вообще полиплодия характерна в основном для человека, а
среди животных встречается крайне редко. При полиплоидии число хромосом в
клетке насчитывается по 69 (триплодие) , а иногда и по 92 (тетраплодие)
хромосомы. Такое изменение ведет практически к 100 % смерти зародыша.
Триплодие имеет не только многочисленные пороки, но и приводит к потере
жизнеспособности. Тетраплодие встречается еще реже, но так же зачастую
приводит к летальному исходу.
Анеуплоидные же мутации приводят к изменению числа хромосом в
кариотипе, некратное гаплоидному набору. В результате такой мутации
возникают осыби с аномальным чилом хромосом. Как и триплодия, анеуплодия
часто приводит к смерти еще на ранних этапах развития зародыша. Причиной же
таких последствий является утрата целой группы сцепления генов в кариотипе.
В цело же, механизм возникновения геномных мутаций связан с
патологией нарушения нормального расхождения хромосом в мейозе, в
результате чего образуются аномальные гаметы, что и ведет к мутации.
Изменения в организме связаны с присутствием генетически разнородных
клеток. Такой процесс называется мозаицизм.
Геномные мутации одни из самых страшных. Они ведут к таким
заболеваниям, как синдром Дауна (трисомия, возникает с частотой 1 больной
на 600 новорожденных), синдром Клайнфельтера и др.
Спонтанные мутации
Мутации, помимо качественных свойств, характеризует и способ
возникновения. Спонтанные (случайные) – мутации, возникающие при нормальных
условиях жизни. Спонтанный процесс зависит от внешних и внутренних факторов
( биологические, химические, физические ). Спонтанные мутации возникают у
человека в соматических и генеративных тканях. Метод определения спонтанных
мутаций основан на том, что у детей появляется доминантный признак, хотя у
его родителей он отсутствует. Проведенное в Дании исследование показали,
что примерно одна из 24000 гамет несет в себе доминантную мутацию. Ученый
же Холдейн рассчитал среднюю вероятность появления спонтанных мутаций,
которая оказалась равна 5*10-5 за поколение. Другой ученый Курт Браун
предложил прямой метод оценки таких мутаций, а именно: число мутаций
разделить на удвоенное количество обследованных индивидов.
Индуцированные мутации
Индуцированный мутагенез – это искусственное получение мутаций с
помощью мутагенов различной природы. Впервые способность ионизирующих
излучений вызывать мутации была обнаружена Г.А. Надсоном и Г.С. Филлиповым.
Затем, проводя обширные исследования, была установлена радиобиологическая
зависимость мутаций. В 1927 году американским ученым Джозефом Мюллером было
доказано, что частота мутаций увеличивается с увеличением дозы воздействия.
В конце сороковых годов открыли существование мощных химических мутагенов,
которые вызывали серьезные повреждения ДНК человека для целого ряда
вирусов. Одним из примеров воздействия мутагенов на человека может служить
эндомитоз – удвоение хромосом с последующим делением центромер, но без
расхождения хромосом.
Таблица. Приблизительная частота мутаций различных генов у человека.
|Характер |Заболевание |Частота мутаций |Число мутаций на 10|
|наследования | | |в 6 гамет |
| |Туберкулезный |8*10-4 |800 |
|Аутосомно– |склероз | | |
|доминантный | | | |
| |Талассемия |4*10-4 |400 |
| |Ретинобластома |2.3*10-5 |23 |
| |Аниридия |5*10-6 |--- |
|Аутосомно- |Альбинизм |2.8*10-5 |28 |
|рецессивный | | | |
| |Цветовая слепота |2.8*10-5 |28 |
| |Ихтиоз |1.1*10-5 |11 |
|Рецессивный |Гемофилия |3.2*10-5 |32 |
Курение вызывает мутацию
Многочисленные статистические данные об увеличении риска развития
злокачественных опухолей в результате курения сигарет подтверждаются
результатами молекулярно-биологических исследований, которые показывают,
что в клетках курильщиков (причем не только в клетках легких) накапливаются
мутации, многие из которых характерны для злокачественных клеток, сообщает
журнал "Терра Медика Нова". До сих пор такие исследования проводились на
взрослых, которые уже имели значительный фон соматических мутаций,
накопленных даже в отсутствие курения. Опубликованы результаты сравнения
генетического материала, полученного из пуповинной крови новорожденных,
родившихся от матерей, которые не подвергались действию табачного дыма, и
матерей, которые, хотя сами не курили, но были вынуждены находиться в
присутствии курильщиков. Такое пассивное курение приводило к тому, что у
новорожденных спектр мутаций сдвигался в сторону делеций, характерных для
детских лейкемий и лимфом.
Мутации передаются через поколения
Английским ученым удалось показать, что мутации, вызванные
радиационным излучением, были обнаружены в третьем поколении лабораторных
мышей.
В статье, опубликованной в последнем номере Proceedings of the
National Academy of Sciences, впервые продемонстрировано наследование
мутаций, вызванных радиацией, от одного поколения к другому.
Предыдущие исследования, проведенные среди семей, пострадавших во
время ядерной бомбардировки Хиросимы и аварии на ЧАЭС, не выявили
наследования мутаций.
В эксперименте ученые подвергали облучению рентгеновскими лучами
группу мышей-самцов двух разных линий. После этого ученые скрещивали
облученных самцов с необлученными самками и определяли уровень мутаций у их
потомства. Было показано, что как дети, так и внуки облученных самцов несли
изменения в последовательности ДНК и демонстрировали более высокий уровень
мутаций по сравнению с мышами дикого типа.
Ученые полагают, что факт наследования мутаций вызывает определенные
опасения, поскольку это может увеличить риск развития рака или других
заболеваний.
У людей генетическое разнообразие гораздо выше, чем у мышей,
используемых в эксперименте. Поэтому то, что наследование мутаций у людей
до сих пор не обнаружено, не означает, что его нет. По мнению ученых,
картирование человеческого генома может помочь обнаружить наследуемые
мутации.
Поделиться925.11.2011 16:05
- член общества

- Откуда: Россия
- Зарегистрирован: 06.08.2011
- Приглашений: 0
- Сообщений: 12562
- Уважение: +282
- Пол: Женский
- Провел на форуме:
4 месяца 9 дней - Последний визит:
11.01.2018 22:50
http://medicalplanet.su/genetica/167.html
Наиболее распространенный тип количественных аномалий хромосом -трисомии и тетрасомии по одной из пар. У живорожденных чаще всего встречаются трисомии по 8, 9, 13, 18, 21 и 22 аутосомам. При возникновении трисомии по другим аутосомам (особенно большим метацентрическим и субметацентрическим), эмбрион оказывается нежизнеспособным и гибнет на ранних сроках внутриутробного развития. Летальный эффект имеют и моносомии по всем аутосомам.
Выделяют два цитогенетических варианта трисомий: транслокационный и регулярный. Первый вариант достаточно редко выступает в качестве этиологического фактора и составляет не более 5% всех случаев трисомий по аутосомам. Транслокационные варианты синдромов хромосомных трисомий могут появляться у потомков носителей сбалансированных хромосомных перестроек (чаще всего, робертсоновских или реципрокных транслокаций и инверсий), а также возникать de novo.
Остальные 95% случаев трисомий по аутосомам представлены регулярными трисомиями. Существует две основные формы регулярных трисомий: полная и мозаичная. В подавляющем большинстве случаев (до 98%) обнаруживаются полные формы, возникновение которых может быть обусловлено, как гаметическими мутациями (нерасхождением или анафазным отставанием хромосомы при мейотическом делении одной единственной гаметы), так и наличием сбалансированных хромосомных перестроек во всех клетках родителей. В редких случаях наследование количественных хромосомных перестроек происходит от родителей, имеющих полную форму трисомий (например, по Х- или 21-хромосоме).
Мозаичные формы трисомий составляют около 2% всех случаев и характеризуются различным соотношением нормальных и трисомных клеточных клонов, которое и определяет вариабельность клинических проявлений.
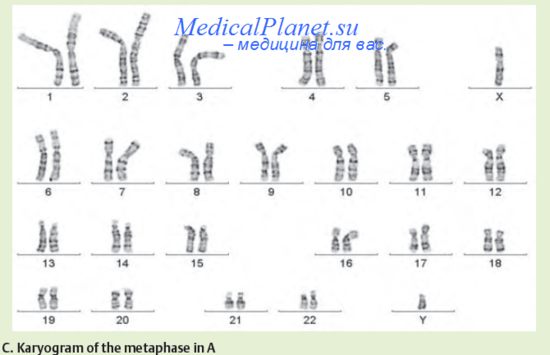
Приводим основные клинико-цитогенетические характеристики трех наиболее распространенных вариантов полных трисомий по аутосомам у человека.Синдром Дауна или трисомия по 21-й хромосоме.
Синдром был впервые описан английским врачом Дж, Дауном в 1866 г., ацитогенетически охарактеризован Ж. Леженом в 1958 г. Это наиболее распространенная хромосомная патология, встречающаяся с частотой 1:700-800 новорожденных. Оба пола поражаются одинаково часто. 95% всех случаев синдрома Дауна представлено регулярной трисомией, возникающей в результате нерасхождения хромосом в мейозе и лишь 5% обусловлено наличием унаследованной от родителей или возникшей de novo робертсоновской транслокацией с вовлечением 21 хромосомы. Наиболее часто (примерно в 80% случаев) нерасхождение хромосом происходит во время первого мейотического деления, причем, в 2/3 случаев в женских половых клетках. Важно отметить, что вероятность нерасхождения значительно возрастает при увеличении возраста женщины. Так, частота рождения детей с синдромом Дауна у 20-летних женщин составляет 1: 1800, у 30-летних - 1: 1000, а у 40-летних - 1: 100 новорожденных.
В подавляющем большинстве случаев (98%) выявляется полная форма синдрома Дауна, и лишь 2% больных имеют мозаичный вариант заболевания.
В настоящее время патогенетические механизмы возникновения клинических симптомов при увеличении хромосомного материала окончательно не расшифрованы. Предполагают, что ключевая роль в возникновении умственной отсталости при синдрома Дауна принадлежит увеличению дозы гена фермента супероксиддисмутазы, локализованного в области q22.3.Клинические симптомы синдрома Дауна очень специфичны и позволяют диагностировать болезнь уже в родильном доме. Больные имеют характерное уплощенное лицо, монголоидный разрез глаз, эпикант, короткий нос с плоской спинкой, макроглоссию, брахицефалию, низко расположенные, как правило, деформированные ушные раковины. Еще одно проявление синдрома Дауна — изменение дерматоглифики: присутствие длинной поперечной складки на ладонях и двух кожных складок на мизинце. Кисти больных обычно короткие и широкие с клинодактилией мизинцев. Пороки сердца отмечаются у 53% больных, пороки развития желудочно-кишечного тракта - у 15% пораженных, аномалии почек выявляются лишь в 6% случаев. Для всех больных характерна мышечная гипотония и снижение роста, у взрослых, как правило, обнаруживается избыточный вес тела. Наиболее важный симптом при синдроме Дауна - олигофрения различной степени тяжести. Дети с синдромом Дауна обычно ласковы, терпеливы, послушны, что в ряде случаев, при использовании специальных обучающих программ приводит к овладению ими навыков самообслуживания, определенной социальной адаптации и даже освоению облегченной школьной программы. Продолжительность жизни больных с синдромом Дауна, как правило, снижена, они редко доживают до 40-летнего возраста, что связано со снижением клеточного и гуморального иммунитета, нарушением процессов репарации ДНК и формированием признаков преждевременного старения в мозге больных. Смерть часто наступает от сердечной недостаточности, интеркуррентных инфекций, а в ряде случаев онкологических заболеваний, прежде всего лейкемии.
Диагностика заболевания осуществляется на основании клинического осмотра и анализа кариотипа больного. В случае выявления регулярной трисомии по 21 -й хромосоме исследование кариотипа родителей больного не проводят. При обнаружении транслокационной формы заболевания этот этап медико-генетического консультирования семьи абсолютно необходим. Профилактика заболевания осуществляется на основании дородовой диагностики, основанной на цитогенетическом и ультразвуковом обследовании плода. Такое обследование необходимо проводить в двух случаях:
1) если беременная женщина старше 35 лет (в связи со значительным увеличением риска возникновения хромосомных перестроек в этом возрасте);
2) если у одного из родителей больного выявлена робертсоновская транслокация с вовлечением 21 хромосомы.
Поделиться1025.11.2011 16:18
- член общества
- Откуда: Россия
- Зарегистрирован: 06.08.2011
- Приглашений: 0
- Сообщений: 12562
- Уважение: +282
- Пол: Женский
- Провел на форуме:
4 месяца 9 дней - Последний визит:
11.01.2018 22:50
http://www.strf.ru/material.aspx?Catalo … d_no=27405
Синдром Дауна – разработка новых методов ранней диагностики
Специалисты ГНЦ РФ ГосНИИ генетики и селекции промышленных микроорганизмов и Медико-генетического научного центра РАМН разрабатывают методику, которая позволяет быстро и без осложнений определить синдром Дауна у плода при сроке беременности 10-15 недель. Метод основан на анализе молекул РНК плода, циркулирующих в крови матери. Работу ученых поддержал РФФИ.Причина синдрома Дауна - лишняя копия 21-й хромосомы или ее фрагмента. Тяжесть этого синдрома и степень проявления разных симптомов (умственной отсталости, сердечно-сосудистой и урогенитальной патологий, аномалий опорно-двигательного аппарата и морфологии черепа) зависят от многих невыясненных факторов, но даже в легких формах синдром Дауна вызывает раннюю инвалидность. В настоящее время пренатальную диагностику проводят, анализируя хромосомный набор клеток плода. Это очень точный метод, но он обладает двумя серьезными недостатками. Чтобы получить материал для исследования, приходится брать пробы околоплодной жидкости или делать биопсию хориона или плаценты. Такое вмешательство в 0,5-2% случаев вызывает прерывание беременности. Кроме того, анализ хромосом плода занимает 2-3 недели, что сокращает возможности раннего аборта, если родители на него решаться.
Московские ученые разрабатывают более щадящий и быстрый метод, доступный для применения в российских лабораториях. Он основан на анализе РНК некоторых генов 21-й хромосомы. Эта РНК циркулирует в крови матери и ее можно отличить от родительской. РНК выделяют из венозной крови. Исследования показали, что кровь и плазму можно хранить на льду в течение 24 ч, и РНК при этом почти не разрушается. Следовательно, у медиков достаточно времени, чтобы доставить образцы из женских консультаций в диагностические лаборатории. Но есть еще реагент TriZol, который позволяет хранить образцы плазмы как минимум полгода.
Сложнее всего оказалось подобрать подходящие для анализа гены. Они должны располагаться на 21-й хромосоме и работать только в той части плаценты, которая содержит клетки плода. Пока что ученые подобрали один подходящий маркер – фрагмент гена PLAC4. К сожалению, одного маркера недостаточно для точной постановки диагноза. Исследователи планируют провести более полный анализ генов хромосомы 21, и выбрать те из них, которые функционируют в соответствующих частях плаценты. Ученые надеются, что объединив данные нескольких десятков маркеров, можно будет уверенно определять синдром Дауна у плода по образцам материнской крови.
Аналогичные работы уже начаты за рубежом, но ученым пока не удалось добиться абсолютной специфичности и высокой чувствительности метода.
Источник информации: Благодатских Е.Г., Серегин Ю.А., Маркова Ж.Г., шилова Н.В., Носиков В.В., Золотухина Т.В. «Возможности использования циркулирующих в крови матери мРНК плацентарного происхождения для диагностики синдрома Дауна у плода». Медицинская генетика, 2009, № 12
Дополнительная информация: Золотухина Татьяна Владимировна, д.б.н., профессор, Медико-генетический научный центр РАМН, Москва; ztv@med-gen.ru;
Серегин Юрий Александрович, к.б.н., ГНЦ РФ ГосНИИ генетики и селекции промышленных микроорганизмов, Москва; seryogin@genetika.ru
ГНЦ РФ ГосНИИ генетики и селекции промышленных микроорганизмов и Медико-генетического научного центра РАМН
Поделиться1120.12.2011 16:31
вроде этого не было
Регистр СД, являющийся составной частью Национального регистра хромосомной патологии, включает цитогенетические, фенотипические и формально-генетические характеристики 953 пациентов (1983–2001 гг. рождения) с СД, подтвержденным методом дифференциального окрашивания хромосом лимфоцитов периферической крови. Цитогенетические данные: Регистр содержит сведения о 840 пациентах с полной трисомией 21, 79 больных с транслокационной формой СД вследствие как спорадических, так и унаследованных робертсоновских транслокаций и других структурных перестроек, 13 случаев комплексных аномалий и 21 пациент с мозаичным кариотипом. Редкие аберрации включали числовые перестройки: 48, ХХУ + 21; структурный дисбаланс: 46, XX, psudic(21;21)(21pter- >21q22::21q22- >pter); 46, XY, der(9) t(9;21)(p24.3; q11.2); 46, XX, dup(21)(q22q11.2); комплексные перестройки, представленные сочетанием полной трисомии 21 с неидентифицированным маркером (48, XY, +21, +mar), с реципрокными транслокациями (47, XX, t(11;21)(q21; q22) mat, +21; 47, XY, t(12;22)(p11.2; q13) pat, +21; 47, XX, t(9;14)(q13; q22) +21) и инверсиями (47, XY, inv(7)(p12q21.1) mat, +21; 47, XX, inv(9)(p11q13), +21). Мозаичные формы включают как типичный (наиболее частый) вариант — 47, XY, +21/46, XY, так и редкие числовые (47, XY, +21/48, XY +21; +21), структурные (46, XX, der (21;21)(q10; q10), +21/46, XX), а также комплексные перестройки (48, XY, +21, +mar/46, XY). Большая часть случаев транслокационного СД обусловлена вновь возникшими аберрациями: удельный вес спорадических мутаций среди информативных семей составил 64,9%. Среди унаследованных форм СД в подавляющем большинстве случаев (17 из 20) заболевание у ребенка явилось следствием сегрегации робертсоновских транслокаций, имеющих место у одного из родителей (такие транслокации были выявлены у 14 матерей и 3 отцов).
Поделиться1220.12.2011 17:05
http://www.krepublishers.com/02-Journal … a-N-Tt.pdf
2010 год про все виды трисомии ,данные исследований, текст надо перевести правда))
Поделиться1320.12.2011 17:15
Хромосомные аномалии в DS:
Стандартный трисомии 21
Дополнительной хромосомы 21 от мейоза нерасхождения или неудачи пар хромосом отделить во время образования гамет присутствует в около 95% людей с синдромом Дауна. [2] Популяционные исследования показывают, что более 90% нерасхождения ошибки, приводящие к трисомии 21 место в яйцеклетку и преимущественно в материнской мейоза I. [3] [4]
Хромосомных кариотипов для стандартных трисомии 21 обозначается как 47, XY, +21 у мужчин и 47, ХХ, +21 у женщин.
Робертсоновских транслокации
Около 4% имеют 46 хромосом, одна из которых является робертсоновских транслокации между 21q и длинного плеча одной из других акроцентрических хромосом (обычно хромосомы 14 или 21). [2] транслокация хромосомы заменяет один из нормальных acrocentrics. Кариотип человека с синдромом Дауна и робертсоновских транслокации между хромосомами 14 и 21 обозначается как 46, XX или XY, дер (14; 21) (q10; q10), +21.
Перемещение DS имеет относительно высокий риск повторения в семьях, когда один из родителей является носителем транслокации. По этим причинам, хромосомные кариотипирование родителей имеет важное значение, прежде чем предоставлять повторения риски при последующих беременностях.
21q21q робертсоновских транслокации является хромосома, состоящая из 2 21-й хромосомы длинные руки. Это бывает редко. В этом случае, все гаметы перевозчика хромосомы содержат 21q21q хромосомы, с двойной дозы хромосоме 21q и отсутствие какой-либо хромосомы 21P генетического материала. Носителем 21q21q транслокация 100% повторение риск рождения ребенка с 21q21q транслокации. [2]
Мозаика
Около 1% из них мозаику, с клеточной популяции, содержащие как нормальные и трисомии 21 кариотип. Фенотип, как считается, мягче, чем трисомия 21, но когнитивные и поведенческие профили меняются.
http://bestpractice.bmj.com/best-practi … ology.html
Поделиться1420.12.2011 17:19
Регулярные трисомии 21 (М: F = 2,21)
47, XY, +21
47, ХХ, +21
Перемещение (M: Ж = 1,43)
46, XX, дер (13; 21) (q10; q10), +21
46, XY, дер (14; 21) (q10; q10), +21
46, XX, дер (14; 21) (q10; q10), +21
6
46, XY, дер (21; 21) (q10; q10), +21
9
46, XX, дер (21; 21) (q10; q10), +21
3
46, XX, дер (21; 21) (q22.3; q22.3), +21
46, XX, дер (21; 22) (q10; q10), +21
46, XX, дер (15; 21) (q10; q10), +21
46, XY, дер (15; 21) (q10; q10), +21
46, XY, idup (21; 21) (q22.3; q22.3), +21
47, XY, т (11; 21) (q13.3; p13), +21
46, XY, инв (9) (QH), дер (21; 21) (q10; q10), 1
+21
Мозаика (M: F = 2,0)
47, XY, +21 / 46, XY
47, ХХ, +21 / 46, XX
46, XY/46, XY, дер (21; 21) (q10; q10), +21 1
47, XY, дер (21; 21) (q10; q10) / 47, XY, +21 1
Неклассические (M: F = 3.50)
47, XY, инв (9) (Qh), +21
47, ХХ, +21 / 48, XXY, +21
46, XY, дер (13; 14) (q10; q10), +21
47, X, инв (Y), +21
47, XX, дер (11; 22) (q13; q11), +21
Поделиться1509.01.2012 00:27
- член общества

- Откуда: Москва
- Зарегистрирован: 08.11.2011
- Приглашений: 0
- Сообщений: 17911
- Уважение: +619
- Пол: Женский
- Провел на форуме:
8 месяцев 27 дней - Последний визит:
23.12.2022 08:26
вот что нашла про СД(я этого не знала)
После 20 лет у 100% пациентов в головном мозге обнаруживают бляшки, характерные для болезни Альцхаймера.
Низкое содержание АФП в сыворотке матери на 14–16 нед беременности (помогает выявить лишь 1/3 случаев)
из этой статьи Кликай
Отредактировано Юлия (09.01.2012 00:30)
Поделиться1610.01.2012 18:49
- член общества
- Откуда: Москва
- Зарегистрирован: 08.11.2011
- Приглашений: 0
- Сообщений: 17911
- Уважение: +619
- Пол: Женский
- Провел на форуме:
8 месяцев 27 дней - Последний визит:
23.12.2022 08:26
Вот что ещё нашла:
Синдром делеции длинного плеча 21 хромосомы (синдром 21 q-). Выделен в отдельную нозологическую группу в 1970 году. Цитогенетически характеризуется частичной делециеи длинного плеча хромосомы 21, а также кольцевой хромосомой. Отмечены мозаичные формы этих хромосомных нарушений. Диагностическими признаками заболевания являются: низкая масса тела при рождении, микроцефалия, антимонголоидный разрез глаз, эпикант, широкая переносица, большиенизко посаженные деформированные уши с расширенным слуховым каналом, микрогнатия. К основным признакамотносятся также сколиоз, клинодактилия, паховая грыжа, косолапость и крипторхизм. Их пороков внутренних органов наблюдаются пороки сердца и патология почек (гидронефроз, удвоение почечных лоханок). Отмечена задержка психомоторного развития. При грубых пороках развития дети умирают в первые недели жизни, хотя описаны больные в возрасте 50 лет.
Синдром кольцевой хромосомы 22. Выделен в самостоятельный синдром в 1972 году. Цитогенетические варианты могутбыть различны: кольцевая хромосома 22, делеции длинного плеча этой хромосомы и мозаичные варианты. В 90% случаев данные хромосомные нарушения являются мутациями de novo. Основными диагностическими признаками заболеванияя вляются: микроцефалия, эпикант, гипертелоризм, крупные выступающие глаза ("глаза лани"), расщелина язычка инеба, густые брови и ресницы. Кроме того, отмечается дисплазия тазобедренных суставов, клинодактилия и синдактилия. Пороки внутренних органов не характерны. Умственная отсталость проявляется как легкой, так и выраженной олигофренией. Больные легко возбудимы, отмечается частая смена настроения, нарушение координации, а также грубое недоразвитие речи, вплоть до ее отсутствия.
Синдром трисомии длинного плеча 14-й хромосомы (синдром 14q+). Описан в 1970 году. Цитогенетически проявляетсякак дупликация участка хромосомы 14, имеются транслокационные формы и изохромосома 14 по длинному плечу. Типичными признаками заболевания являются: микроцефалия, луковицеобразный нос, тонкая верхняя губа, микростомия, опущенные углы рта, низкорасположенные ротированные назад ушные раковины, короткая шея. Характерно неправильное расположение пальцев на руке, клинодактилия, косолапость. Из внутренних пороков описана патология сердечно-сосудистой системы. Дети резко отстают в психомоторном и физическом развитии.
Поделиться1715.01.2012 08:48
корявый перевод с иврита
Хромосомные аберрации
А. Типы хромосомных аберраций
Правильное количество хромосом и их правильная структура является критически важным для роста и нормального развития плода. Иногда дети рождаются с изменением числа хромосом. Излишки генетический багаж или пропавших без вести это может привести к росту и развитию ребенка. Режим в ячейке или живое существо которого избыток хромосом или отсутствующие хромосомы называется Anaifloaidih (анеуплоидии), а не нормальным пониманием Aifloaidih (euploidy), а в ситуации, или живое существо более массив одного или нескольких хромосом, называется Polifloaidih как Trifloaidih или tetraploidy.
На самом деле хромосомные аберрации являются общими. От 15% до 20% беременностей заканчивается абортом известно населения и половина из них из-за хромосомных аберраций.
Хромосомные аберрации включают в себя: изменение числа хромосом (Polifloaidih и Anafloaidih) и их структуры хромосом изменения, которые мы будем подробно в третьей главе.
В. Изменения числа хромосомИзменения числа хромосом, связанных с потерей хромосом или наличием внештатных хромосом, и бороться наличие дополнительных фрагментов хромосом от неизвестного источника. Следующие типы изменений.
1. Polifloaidih - ПолиплоидияAifloaidi ячейка содержит нормальное количество массивов. Например, нормальные человеческие клетки (остальные примеры относятся к человеческому роду) содержит два набора т.е. 23 пары хромосом. Нормальная клетка тела содержит два набора по 23 хромосомы называют диплоидные клетки. Секс клетки (сперматозоиды или яйцеклетки) содержит одно значение, и поэтому она называется клетка Hfloaidi (греч. haploos = один, простой).
Trifloaidih ненормальна ситуация, в которой Есть три набора хромосом, а в общей сложности 69 хромосом вместо 46. Tetraploidy это ненормальное состояние, в котором Есть четыре набора хромосом вместо двух.
Два из этих ненормальных ситуаций, ведущих ранний аборт, тем не менее, небольшое количество плодов с Trifloaidih дожить до последнего триместра беременности.
2. Monoseomih - моносомияMonoseomih это наследственное заболевание, в которых только одна хромосома Humlogy отсутствие супруга, если такая ячейка в клетке человека содержит 45 хромосом вместо 46. Большинство Hhtabreuot с Monoseomih не выжить имплантации этап в утробе матери. Это объясняет, почему Monoseomih как правило, не наблюдается даже аборты на ранних сроках. Отсутствие хромосомы как правило, имеет более сильное воздействие на развитие плода, чем избыток хромосом.
За исключением нескольких редких случаях Monoseomih 21, Monoseomih женской Х-хромосоме секс-смотрел беременности. 99% процентов Mhhtabreuot с Monoseomih X преждевременной делают аборты. Остальные девушки в конечном итоге при рождении с синдромом Тернера (синдром Тернера). Женщины с синдромом Тернера имеют наружных женских половых органов, но не имеют яичников и периодов, и поэтому они не являются плодородными. Эти женщины являются низкими. Подавляющее большинство психически нормальным.
Нормальная женщина представляется в следующем виде 46 XX в то время как женщины с синдромом Тернера около 45 X0.
3. Трисомия - ТрисомияТрисомия это условие не использовать его правильно в настоящее время три (вместо двух гомологичных хромосом) копии частности хромосомы и обнаружили в общей сложности 47 хромосом вместо 46. С другой стороны, Disomih является нормальной стандартной ячейки, гомологичных хромосом пара. Трисомия является общим и оценивается в 20% Hhtabreuot известна, и она движется от 2% до Mhhtabreuot среди молодых женщин в возрасте от 25 до 35% Mhhtabreuot у женщин старше 40 лет.
Синдром Дауна является самой распространенной симптомы из-за трисомии. Синдром Дауна является частично вызвано наличием дополнительной копии хромосомы 21, так что синдром Дауна называется также трисомии 21. Упоминание, хромосомы пронумерованы в соответствии с размером кариотип. Кариотип синдромом Дауна был написан так, что 47 ХХ +21, женщина или мужчина, 47 XY +21. Восемьдесят процентов Mhtabreuot с трисомии 21 завершится в начале выкидыша.
Кроме синдрома Дауна, трисомия 21, Есть дальнейшего их причины трисомии синдромов. Adaord синдром (синдром Эдварда) от трисомии 18 и патио синдром (синдром Патау) трисомии 13 возникла.
Трисомии 13 и трисомии 18 хромосомные аберрации тяжелой, смертельной, как правило, приводит к смерти в раннем детском возрасте, часто в первый год. Большинство Hhtabreuot с трисомии 13 или 18 заканчивается абортом ранее. В общем, трисомии хромосом Aotozumliim (без половых хромосом) приводит к ранний аборт.Трисомии хромосомы влияет на секс меньше на рост и развитие ребенка, а иногда и людей с лишней хромосомы секс может быть без особых симптомов. Примеры половых хромосом трисомии являются: XXX, XYY, XXY. Клайнфельтера синдром (синдром Клайнфельтера) является его кариотип трисомия 47, XXY. Человек с этим синдромом характерны структуры высокий, тонкий корпус, маленькие яички, отсутствие семян, увеличение груди и отсутствие шерсти на теле и лице.
4. Хромосомные маркер - маркер хромосомХромосомные маркер сегмента хромосомы, как правило, неизвестного происхождения. Если известно, из-за изменения в хромосоме подавлено. Эффект от этого дополнительный генетический груз на рост и развитие зависит от хромосомы участие и количество клеточных проходы.
C. Изменения структуры хромосомСуществует различие между точечных мутаций в ДНК и хромосомы структурные изменения, которые можно смотреть в микроскоп. Вот изменения (мутации), которые видны в микроскоп, и все Bbshinweim больших и разных сегментов.
1. Пропустить - Удаление
Потеря хромосомы сегменте2. Дублирование - дублированию
Другая копия гена показывает тот же порядок на хромосоме.3. Flip - Инверсия
Повторное расположение генов в хромосоме.4. Водоизмещение - транслокации
Замена сегментов между двух гомологичных хромосом.5. Хромосома круг - кольцо хромосомы
Обрезать концы хромосом, а затем соединение заканчивается.6. Номер - вставка
Добавление часть хромосомы, где это не должно быть.7. Усиление - Усиление
Htoosfot трио повторяется нуклеотидных химический процесс, при передаче генов от родителей к потомству.
Поделиться1815.01.2012 08:50
тот же корявый перевод с иврита
Перемещение мутаций - транслокации
А. Что такое перемещение?
Перемещение мутации очень распространенных хромосомных аберраций у людей. Является ли одной из причин синдрома Дауна, и происхождение синдрома Fradr - и Вилли. Кроме потери рациональности он может поставить под угрозу способность плодородия и вызывают рак. В большинстве случаев перемещения мутация изменением структуры хромосом, и меньшинство случаев имеется отклонение от нормального числа хромосом. Проще говоря, вытеснение мутации является замена сегментов между двумя гомологичными хромосомами, а не о той же паре. Иллюстрация ниже, слева, две пары гомологичных хромосом в нормальных 5 и 8. На правой стороне, две гомологичные хромосомы обмен сегментами после взаимной.
Последнее перемещение хромосом может сильно отличаться от первоначальной длины, и это зависит от длины секций Н "htocim". Смотрите кариотип взаимное смещение между хромосомы 5 и 8, обратите внимание, что случилось с хромосомой 5 правых.
Термин перемещения (перемещения) является переводом на английский язык слово "Translokatzih", что означает изменение места, переезжая из одного места в другое. Она включает в себя иврите разделения, разрыв и переход от одного места в другое. Действительно, мутации хромосом сегменте смещение перерывов и соединяется на различные хромосомы ( см. рисунок ). Письменные везде смещение взаимное смещение означает, если не указано иное.
В. Перемещение симметричный и несимметричныйПеремещение мутации не могут быть сбалансированы (несимметричный), если ячейка является дисбаланс или избыток генетического материала или сбалансированным (сбалансированный), если не в недостатке или избытке создан ( см. рис смещение сбалансированный). Кроме машины сбалансированной вопрос перемещения подняли, как будто он мог бы оставить здоровое потомство несбалансированной перемещения и имеет серьезные последствия для здоровья. Рисунке слева ясно дает понять, как тайский воспроизведение носителей, образующихся в процессе мейоза, несбалансированное вопросы перемещения. Хотя отдельные сбалансированные вопросу смещение его клетки, он не проявляет никаких проблем со здоровьем, кроме бесплодия: снижение рождаемости (катапульты Htabreuot), гибель плода (отсутствие имплантации или выкидыши) и тяжелых дефектов в потомстве.
Действительно, смещение сбалансированным или избытка без потери генетического материала, но может быть потеря генетической информации по нескольким причинам. С одной стороны, точки останова внутри гена приводит к сегментации гена является необходимым и публикации действие Cbtsmont Fradr - и Вилли. Еще одна причина, вставка разделов месте приводит к перестройке генов и нарушения активности генов. Например, размещение двух удаленных генов рядом друг с другом может повлиять на управление одним геном и вызывать рак.
В следующих главах будет дано объяснение два типа смещения. Взаимное перемещение и смещение в памяти Robertsonein.
C. Что такое взаимное смещение?Взаимное смещение (взаимные транслокации) является перемещение из одного или более части одной хромосомы к хромосоме двух, что не является его супруга, а другой один или несколько разделе второй хромосоме, не обязаны быть в тот же порядок величины, просто подключите разрыв точки первой хромосоме. Помимо взаимных перемещений между хромосомой 11 по 22 хромосоме, в большинстве Hhtkot взаимности создаются случайным образом, и не повторяются знакомые форматы. Распространенность взаимное смещение у людей 1:625.
Смотрите кариотип сбалансированный смещение, которое происходит между хромосоме 5 хромосоме 8. Обратите внимание, что значительная часть длинного плеча хромосомы 5 (право) заменена небольшой части короткого плеча хромосомы 8 (справа).
См. иллюстрацию слева показывает иллюстрации мутаций перемещения, как показано на кариотип , хромосома 5 хромосоме 8. Эта мутация Tsoml следующим образом: 46, XY, т (5; 8) (q31.1; p23.1)
Объяснение:
46 - это количество хромосом. Никаких изменений в число хромосом.
XY - представляет половых хромосом. Это мужской кариотип.
(8, 5) т - представляет собой взаимное смещение между хромосоме 5 хромосоме 8;
(Q31.1; p23.1) переломный момент на длинном плече хромосомы 5 представлена q31.1 и p23.1 представляет переломный момент короткого плеча хромосомы 8.Посмотрел на следующий кариотип и видел, как трудно сказать, смещение символизировал следующим образом:
46, XY, т (2, 15) (p11.2; q11.2). Тщательное наблюдение, сравнивая следы хромосоме 2 его права гомологии, то можно увидеть, что они похожи, как ожидалось. Смотрите иллюстрацию , которая показывает взаимное смещение между очень большой сегмент небольших плеча хромосомы 2 целых плече хромосомы 15.
Д.. Как взаимное смещение происходит?Взаимное смещение в большинстве случаев происходит при случайном процессе создания половых клеток (сперматозоидов или яйцеклеток, что). В большинстве случаев это происходит сразу после завершения Hfrofazh в первом дивизионе сокращения (мейоза I). На Hfrofazh является дублирование хромосом и хромосомы Hhumologyim цепляясь друг друга видеть медленное анимации . Первый этап разделения сокращения случайного распределения Hhumologyim хромосомы, по-прежнему цепляются друг к другу, как тайский - девочка. Создать Тайский - Размножение необходимой нормативной и точное разделение хромосом первого мейоза, и надлежащего разделения Hcrumtidot мейоз II. Обеспечивает надлежащее клетки потомок разделения содержат точные копии своих родителей. Несмотря на замечательный механизм разделения, негры могут быть вызваны из-за их хрупкости и Dbicotm хромосом. Когда два негомологичных хромосом перерыва три возможных исхода:
Реабилитация - подключить сломанной сегментов, которые предела.
Отсутствие мутаций - раздел прервался и будут потеряны во время разделения хромосом.
Взаимное смещение - каждая секция разрывая переломный момент подключается к второй хромосоме.Подключение разделов неуместны могут возникнуть во время движения хромосом мнимой экватору клетки и стабилизации его гомологии с хромосомой и в оппозиции тянуть их к полюсам.
. Что такое смещение в результате чего в комнате?Термин появляется в литературе инсерционного транслокации. Намерение состоит в несбалансированной мутации номер (см. рисунок), в результате перемещения между двумя хромосомами, которые не Humlogym.
А. Что такое смещение рекомбинирующих?
Как уже упоминалось выше, перемещение между двумя частями заменяются не-гомологичных хромосом. Нам не следует путать с подкачки приводит к образованию новых комбинаций генов (рекомбинации).
Своп механизм взаимной замены параллельных отрезков между двумя гомологичными хромосомами или, скорее, между Crumtidot являются сестрами. Замена очень распространена и является наиболее характерных черт мейоза. Замена является одним из механизмов, ответственных за генетические различия между потомством и потомством предыдущих поколений.
Перегруппировка происходит во время создания Тайский - разведение, в течение первых Hfrofazh и заканчивается в начале первого Hanfazh. На первый Hfrofazh каждой хромосомы удвоились и построена из двух соединенных между Btzntrumr сестры Crumtidot. На данный момент две хромосомы Hhumologyim (каждый с двумя Crumtidot) подход и цепляются друг к другу и создать Ttzlovot. Приверженность и Httzlovot того, чтобы разорвать и липкость, которые являются видными характеристики ДНК секли, позволяют подкачки. Неоднозначным благом, если условия позволяют нежелательных изменений в число хромосом, а иногда, как в перемещения.
М. Злокачественные новообразования и взаимное смещениеВ данной главе представлены два примера того, злокачественные заболевания, мутации причиной перемещения. Четкая взаимосвязь между перемещением и злокачественное заболевание было впервые описано в 1960 году после обнаружения небольшой хромосомы в клетках больных частности хронический миелолейкоз. Это хромосома называется «филадельфийской хромосомой», название Вашего города был и остается продуктом взаимного перемещения между Crumzum 9 Llachrumzum 22. См. графический , как ABL гена, расположенного на хромосоме 9, соединяющий BCR сада, расположенного на хромосоме 22. Хотя все генетический материал оказывается (для некоторых пациентов Есть Hshmtim последовательностях из хромосомы 9), но генетическая информация изменилась, потому что это сделало новый ген BCR-ABL филадельфийской хромосомой отвечают за выработку фермента тирозина - киназы играют важную роль для замачивания болезни Хронический миелолейкоз (хронический миелолейкоз. , CML), также известный как хронического миелолейкоза является раком крови характеризуется неконтролируемым распространением белых кровяных клеток, которые вырабатываются в костном мозге, и это взять на себя костный мозг и не допустить его производству здоровых клеток крови, которые. Рост процессов, роста и пролиферации клеток в целом, и особенно злокачественных клеток, основанную на фермент тирозин активации - киназы, и Galibk ингибирует этот фермент. Galibk наркотиками (Гливек) и другое название иматиниба (иматиниб) работает, блокируя (ингибирующие) сигналов между клетками, что рак, а также предотвращение ряд химических реакций, которые заставляют клетку распространять и делиться.
За годы дополнительные отчеты на других перемещенных лиц, которые вызывают других злокачественных заболеваний. Исследователи обнаружили, что в некоторых случаях Hhtkot, свой переломный момент находится вблизи хромосом прото - онкогена. Как правило, прото - онкогена является ген, который не является активным или деятельность которых контролируется, но в результате мутации, происходят изменения в окружении сада, и он мог «найти себя» в других условиях контроля. В результате, операция идет наперекосяк, и она становится онкоген. Oncogene содержащих клеточных размножается бесконтрольно. Например, в результате перемещения из хромосомы 8 по хромосоме 14 суб-прото - онкогена с-Myc, который находится на хромосоме 8, позиции и перемещается в раздел хромосомы 14, который обычно подстегивает образование антител. Изменение места становится прото - онкогена с-Myc онкоген. В результате развития рака, известного как лимфома имя Borkit (лимфома Беркитта). Этот тип рака является рак тканей Hlimfoaidit ответственность перед лимфоцитов и антител. При этом заболевании, Кристиан В-лимфоцитов, белых кровяных клеток, которые вырабатывают антитела, в неконтролируемым образом.
Crumzomim 4 и 8, как правило, ее где-нибудь рядом друг с другом, а белые клетки крови, но не в другие типы клеток. Что и объясняет относительно высокую вероятность мутации смещение именно этого типа клеток. В 85% таких крови Неходжкинская лимфома спит перемещения между хромосомами 14 и 18 происходит во время Hayntrfzh и когда клетка делится в митоз. Это смещение приводит к неконтролируемому работу определенных генов.
Поделиться1923.01.2012 20:01
- член общества
- Откуда: Москва
- Зарегистрирован: 08.11.2011
- Приглашений: 0
- Сообщений: 17911
- Уважение: +619
- Пол: Женский
- Провел на форуме:
8 месяцев 27 дней - Последний визит:
23.12.2022 08:26
Гаметогенез – это процесс образования половых клеток. Протекает он в половых железах – гонадах (в яичниках у самок и в семенниках у самцов). Гаметоге-нез в организме женской особи сводится к образованию женских половых клеток (яйцеклеток) и носит название овогенеза. У особей мужского пола возникают мужские половые клетки (сперматозоиды), процесс образования которых называется сперматогенезом.
Стадии гаметогенеза
1. Стадия размножения. Клетки, из которых в последующем образуются мужские и женские гаметы, называются сперматогониями и овогониями соответственно. Они несут диплоидныйнабор хромосом 2n2c. Первичные половые клетки многократно делятся митозом, в результате чего их количество существенно возрастает. Сперматогонии размножаются в течение всего репродуктивного периода в мужском организме. Размножение овогоний происходит в эмбриональном периоде.
-----------------------------------------------------------------------------------------------------------------------
Девочки,'это что означает?Что хромосомный набор яйцеклеток формируется у женщин,когда они ещё в утробе матери?То корявые яйцеклетки у нас сформировались ещё когда мы были зародышами?
Отредактировано Огонёк (23.01.2012 20:02)
Поделиться2023.01.2012 21:07
- Администратор

- Зарегистрирован: 05.10.2011
- Приглашений: 0
- Сообщений: 5554
- Уважение: +260
- Пол: Мужской
- Провел на форуме:
1 месяц 9 дней - Последний визит:
18.01.2026 19:15
нет
Девочки,'это что означает?Что хромосомный набор яйцеклеток формируется у женщин,когда они ещё в утробе матери?То корявые яйцеклетки у нас сформировались ещё когда мы были зародышами?
нет. овогонии размножились в эмбриональном периоде. то есть заложилось количество будущих яйцеклеток.
http://afonin-59-bio.narod.ru/2_heredit … 06_gsg.htm
http://ru.wikipedia.org/wiki/Мейоз